Carbon
Contents
-
-
-
-
-
-
-
-
-
-
-
Introduction
Carbon is the lightest member of the IVA family of the periodic table, atomic number 6, electronic configuration 1s22s22p2. Its first ionization potential is 11.26V. Its naturally-occurring stable isotopes are of mass 12 (98.89%) and 13 (1.11%). C14 is produced by cosmic rays in the atmosphere at a constant rate, and decays to stable N14 by β- emission with a half life of about 5730 years. A living sample of wood, say, has the equilibrium amount of C14. When it dies, the radiocarbon decays steadily, and the ratio of C14 to C12 declines to half in 5730 years, and so on. By measuring this ratio, the time when the wood was living can be estimated. This method of dating was worked out by Willard Libby in 1947, and is called "carbon dating."
The atomic weight of C12 = 12.0000 was established by the IUPAC in 1961 as the standard of atomic weights. Previously, the naturally occurring isotopic mix of oxygen was taken as 16.0000. This makes the atomic weight of natural carbon 12.010.
Carbon is the 19th most abundant element in the earth's crust, at 0.027%. It is found as carbon dioxide, carbonate rocks, and living matter. It occurs in elemental form in small amounts, as graphite and diamond. The properties of these forms of carbon, which are very important, are discussed in separate sections. The name is derived from the Latin carbo, carbonis, "charcoal."
Like silicon, carbon does not form ionic bonds. Unlike silicon, it forms covalent bonds with other carbon atoms, which can share two, four or six electrons to make single, double or triple bonds, and also single or multiple bonds with nitrogen, oxygen, sulphur and phosphorus. Carbon single bonds are usually sp3 hybrid bonds pointing in tetrahedral directions, as in silicon. The angle between these bonds is 109° 28'. It satisfies its valences with hydrogen rather than with oxygen, as silicon does. This means that carbon can form an extremely wide range of compounds so that it has a chemistry of its own, organic chemistry. Only such a rich treasury of compounds can support the demands of life, so carbon compounds are the basis of all life. These compounds will not be discussed in this article.
We will discuss the elemental forms of carbon and their applications. Carbon is also at the center of the fuels that support the present economy of the world, and fuels will receive close attention. All the curious and unusual uses of carbon that I can find will receive more attention than is their due, as well as the modern topics of buckyballs and nanotubes. Continued reflection turns up traces of carbon in the most varied applications, and these will be reported as long as space is available. We will see once again how important structure is to properties. Carbon is not black, or transparent, or soft, or hard, or a conductor, or a nonconductor--it all depends on structure.
Diamond
 Our word diamond comes from the Latin adámas, adámantis, which is itself a Greek word, adamas, adamantos, meaning in these languages "hard steel," steel that had been heated red-hot and quenched to give a hard, brittle edge or surface. The diamond was a rare gem in the classical Mediterranean, coming from exotic India, also a source of steel and other curiosities. Pliny refers to the diamond as "adamas" as expressing its hardness, the property which gives it its industrial importance. The classical world treasured colored gems more than the clear diamond or quartz, but at the present day the sparkle and artificial costliness of the diamond put it in first place by far. An octahedral diamond crystal is shown in the figure (© Amethyst Galleries).
Our word diamond comes from the Latin adámas, adámantis, which is itself a Greek word, adamas, adamantos, meaning in these languages "hard steel," steel that had been heated red-hot and quenched to give a hard, brittle edge or surface. The diamond was a rare gem in the classical Mediterranean, coming from exotic India, also a source of steel and other curiosities. Pliny refers to the diamond as "adamas" as expressing its hardness, the property which gives it its industrial importance. The classical world treasured colored gems more than the clear diamond or quartz, but at the present day the sparkle and artificial costliness of the diamond put it in first place by far. An octahedral diamond crystal is shown in the figure (© Amethyst Galleries).
Until 1955, all diamonds were natural, though attempts had been made to create them since the beginning of the century. In that year, the long search for artificial diamonds was finally successful, and small industrial-quality diamonds were made from graphite through high temperatures and pressures. In 1970, larger, gem-quality diamonds were artificially made, but they are more costly than natural ones. In the next decade, Russian scientists discovered how to deposit diamond from the gas phase to make hard coatings. Artificial diamonds now command a good fraction of the market for industrial diamonds.
The diamond crystal is made from carbon tetrahedra, just as crystalline silicon is made from silicon tetrahedra. Pauling gives a free-fusion stereogram of the structure. The single bonds are 0.154 nm long. It is a face-centered cubic crystal with lattice constant 0.356 nm, giving a density of 3.51 g/cc, a rather high value for a nonmetallic substance. The pure crystal is transparent, with index of refraction 2.465 at 397 nm, 2.427 at 527 nm, 2.417 at 589 nm, 2.408 at 670 nm, and 2.402 at 763 nm. This difference of 0.063 between UV and IR makes diamond three times as dispersive as a typical glass. Diamond is transparent to X-rays, unlike glass, and can be recognized by this property. Diamond is usually luminescent in UV light, due to impurities that are otherwise not apparent. The high band gap, 5.33 eV, means that diamond should be transparent in the UV to 233 nm. Its specific heat is low, 1.449 cal/g-K, as is its thermal expansion, 0.9 x 10-6 per °C. Its electrical conductivity is extremely low, 2 x 10-21 μS/cm, but its thermal conductivity is high, 5.54 cal/s-cm-K. This high thermal conductivity gives diamond a cold feel, like quartz, but unlike glass. The only substance comparable to diamond in hardness is cubic boron nitride (discovered in 1957). In the Mohs scale, it is 10, but is much harder than the next competitor, corundum (Al2O3) at 9. This makes diamond the ultimate abrasive, able to grind and cut all others. The cubic crystal of diamond cleaves perfectly perpendicular to a 3-fold axis, or parallel to the faces of an octahedron. At a bright red heat, about 870°C, diamond burns to carbon dioxide. In the absence of oxygen, diamond sublimes around 3500°C. A perfect diamond is a single giant molecule, held together by covalent bonds.
The industrial uses of diamond depend only on its hardness; its gem uses depend on its unusually large index of refraction and dispersion. About 80% of diamonds go to industrial uses, 20% to gemstones. A gemstone, or cuttable must be transparent, free of bubbles, inclusions or visible flaws, light in color, and of sufficient size. A fine industrial diamond may be dark brown, but otherwise of gem quality. These diamonds are used for wire-drawing dies of the best quality. Bort represents 99% of industrial diamond. It may be black, have flaws or inclusions, and small in size, but will still be an excellent abrasive. Bort is used for diamond drills, and for diamond wheels. A typical drill diamond may be 20 to the carat. About 1% is composed of microcrystalline aggregates that are very hard and do not cleave, called carbonado, or the spherical aggregates called ballas, most of which comes from Brazil. Its density is 3.15-3.29 g/cc. The three principal industrial uses of diamond are as a general abrasive, for making drills, and as wire-drawing dies. In 1957, the price of industrial diamond was about $4 a carat.
The weight of a diamond is given in metric carats of 0.200 g. The traditional carat varied from 0.197 to 0.207 g in different places. The international carat of 1871 was 0.205 g, and the metric carat was adopted in the U. S. in 1913. The largest diamond ever found, the Cullinan (1905), weighed a little over 3000 ct. If a diamond were a sphere of radius r, then r = 2.39C1/3 mm, where C is the weight in carats. A 1000 ct diamond would be about 2" in diameter, a 100 ct 1", a 10 ct 1/2". You can use this to estimate the weights of any diamonds you may have around. The largest diamond found in the U.S., in Pike County, Arkansas (1906) was 40.22 ct, and the Punch Jones diamond found in Petersburg, W.Va. in 1943 was 34.46 ct. A 100 ct diamond can be considered exceptionally large and valuable, used only in royal jewels. A 10 ct diamond is large enough to be vulgarly displayed and generally admired.
A diamond for gem use is cut to display its clarity and brilliance. The most common cuts are derived from the brilliant cut, shown at the right, which came into use around 1700. It is based on an 8-fold symmetry, and the number of facets is a multiple of 8. The basic brilliant cut has 7 x 8 = 56 facets, plus the top face or table and small bottom face or culet, for 58 facets in all. Each facet has a name; typical names are shown. A major gem is cut from an octahedral crystal as shown, with its girdle corresponding to the widest part of the octahedron. About 50% of the crystal is used. The unused bits can be used to make smaller gems, called melee. None of the natural crystal is wasted. The height of the gem is about 0.6 of the diameter of the girdle. The upper part, or crown is about half as high as the lower part, or pavilion. The purpose of the cut is to return light entering the crown approximately in the direction from which it came, without loss. About 17% of the incident light is reflected at the surface, much more than the usual 4% at a glass surface. White light is dispersed at the final refraction as the light exits the crown; the angular spread of the spectrum is about 2°. This causes color at the boundaries of the reflected areas of light, which are broken up by the many facets. Colorless gem diamonds are preferred, but the rare blue diamonds are treasured. Most colored diamonds are yellow. Though this can be a beautiful color, yellow diamonds are depreciated.
Since diamond is the hardest of all materials, it would seem impossible to cut and smooth the facets on a gem. There must be a trick, and this is it: the hardness of a diamond surface depends on direction. There are hard directions, and soft directions. Though there is not much difference, it is sufficient that the soft direction can be abraded by the hard direction. This must be taken into account even in sawing diamond by diamond, which is an arduous process. The facets of a gem are polished in their soft directions by diamond oriented in the hard direction. Cleaving is the only easy way to split a diamond, and here the slightest error can ruin the work. As might be evident, all this requires much skill and knowledge, so that diamond cutting is a special branch of gem work, distinguished from the less exalted skills of gem cutters and lapidaries, who have diamonds at their disposal and do not have to fret about hard and soft directions.
Natural diamonds were made 1-3 billion years ago in the earth's mantle, at a depth where the pressure was sufficient, say 55,000 atmospheres, and the temperature not too high, perhaps a little over 1000°C. These conditions obtain at a depth of about 150 km below continents, or 200 km below ocean crust. The rock there is peridotite, distinguished as Lherzolite or Harzburgite, composed of 60-90% olivine and 10-40% orthopyroxene, or else eclogite, 50% red garnet and 50% green clinopyroxene, with more iron and aluminium. A diamond begins as a tiny seed, perhaps some garnet, or iron sulphide or carbide. Carbon then grows around it in monomolecular layers, taking millions of years, which often results in well-formed euhedral crystals: octahedrons, cubes, cuboctahedrons, dodecahedrons with rhombohedral faces, or the twinned crystals called macles. The carbon in perioditic diamonds seems to be mantle carbon, while eclogitic diamonds contain carbon with an isotopic ratio typical of organic carbon. This carbon probably was pulled during plate subduction, since eclogitic diamonds were formed mainly in old subduction regions.
These diamonds have been carried to the surface in a few places by violent eruptions caused by the release of active fluids, probably mainly water, in the hot rocks of the upper mantle. The combination of these fluids and the perioditic and eclogitic rocks corrode their way toward the surface and regions of lower pressure, creating dikes and sills and eventually breaking out in hot, turbulent jets that reach the surface in carrot-shaped pipes called diatremes, carrying with them fragments of mantle rocks and rocks dislodged in their ascent, all in a hot, fluidized mass. Most of these eruptions have occurred in the last billion years, typically in the lower Palaeozoic (Devonian), but apparently none since the Cretaceous. The chaotic filling of a diatreme is the soft, easily eroded rock called Kimberlite or Lamproite, typically enriched in potassium, which is otherwise quite rare in such rocks. This is not the only way for diamonds to reach the surface, but it is the principal way, and all commercial diamonds come this way. These diatremes occur in stable craton regions of ancient rocks (because the diamonds were made beneath such regions when they were more active). They can be a half-mile in diameter, and are not rare.
Erosion of the kimberlite pipes frees the heavy diamonds from the matrix, and they join the bed load of streams that drain the area, winding up in alluvial deposits that may have been worked more than once. Diamonds, of course, are extremely durable. All ancient diamonds were found in alluvial deposits, as in India or Burma, and this remains a freqent means of discovering diamond deposits today. Few large diamonds occur in these deposits, but there is a large amount of medium and small crystals. The diamonds have an affinity for petroleum jelly, which easily entangles them. Alluvial diamonds were discovered in western Africa in the 19th century, notably in Congo, Ghana, Ivory Coast, Sierra Leone and other places around the Gulf of Guinea, and today support the economies and revolutions of these impecunious kleptocracies. All you need to mine alluvial diamonds is a shallow metal pan and some persistence.
Diamonds were discovered in southern Africa in 1867, when a 20 ct stone was picked up off the ground by a child. Soon after, the primary deposits of diamond were found in the vicinity of Kimberley, which gave its name to kimberlite. Although these deposits do not produce the majority of diamonds, they are impressive and famous for the scale and intensity of mining. Fresh kimberlite is also called blue earth. Weathered, it becomes yellow earth, from which the diamonds are easily separated. 1600 lb of blue earth may yield only 1/4 ct, 50 mg, of diamond. If they were not so valuable, they would be totally neglected. The Kimberley pipes are Cretaceous in age, among the youngest known.
The blue earth was mined originally in open pits, but then underground as the workings became deeper. At one time, the blue earth was allowed to weather so it was easier to crush and sort, but now the blue earth is processed at once, saving the cost of the inventory lying in the sun. The crushed blue earth is washed to separate the lighter gangue. The remaining 1% is then further enriched by jigging, as with gold, but the diamonds are not nearly as heavy as gold, so it is a more difficult process. Then the residue is greased, or allowed to move down a trough coated with petroleum jelly, to which the diamonds adhere much more strongly than the gangue does. This process was introduced in 1896. Then the greasy diamonds are cleaned, removing the petroleum jelly and leaving the product to be graded.
For many years, South Africa had a near-monopoly of diamonds, because of the scale of their operations. However, recent discoveries have pushed South Africa out of the limelight. Most of the industrial diamonds, perhaps 70%, used to come from the southern Congo. The Congo deposits are in part due to a Triassic pipe at Bakwanga, whose erosion left diamonds in the Karroo gravels. Botswana is now a larger diamond producer than South Africa. In 1960, Devonian kimberlite pipes were found at Zarnitsa, in the Yakut A.S.S.R., now the Republic of Sakha, in deepest Siberia. These deposits, which include gem-grade diamonds, have made Russia one of the largest diamond producers. Probably the most significant new discovery came in 1981 in far northern Western Australia, at the Argyle mines in the far northeastern corner near Wyndham, and at Ellendale near Fitzroy Crossing. Exploration began in 1969 after 9 diamonds were found in the Leonard River. Full scale exploitation began in December 1985. The mainly eclogitic diamonds are in lamproite, and include rare and valuable pink diamonds. Of the Argyle diamonds, 55% are industrial, 45% near-gem, and 5% gem quality, of which 95% are brown, 4% colorless or grey, and only 1% the valuable pink or red. There has been some effort to sell the brown diamonds by giving them attractive names.
Near Murfreesboro, Arkansas, J. W. Huddleston picked up some diamonds in Prairie Creek in 1896. One was cut to a 3.0 ct white diamond, and the other to a 1.5 ct yellow, in New York, which made the site, Crater of Diamonds, where the diamonds were found in a lamproite pipe. The largest found, in 1906, was the Uncle Sam, 40 ct, referenced above. This was never a commercial mine, and is now a state park, where you can pan for diamonds for a small fee. The source of the Punch Jones diamond found in West Virginia seems to be unknown, and no more have turned up.
Kimberlite or lamproite pipes from late Proterozoic to early Devonian have been found in metamorphosed sediments and volcanic rocks of early Proterozoic age in the State Line field between Laramie, Wyoming and Ft. Collins, Colorado, and in the Iron Mountain field northeast of Laramie. At least 40 diatremes have been located. The search for these began in 1975, and a 28.3 ct gem diamond has already been found. Serious mining was supposed to have begun in 1996.
Some time ago, a diamond was found in glacial till in Wisconsin, that had been carried south by Pleistocene continental glaciers from somewhere in Canada. In 1981 some diamonds, or diamond indicators, had been found in the MacKenzie River, so there was some prospecting by large diamond-mining concerns, but nothing was found, and they sold out to one of their contractors, Chuck Fipke. Fipke finally found what he knew was there in 1985, under Lac de Gras, 300 miles northeast of Yellowknife. This finding was made with the scientific assistance of Professor John Gurney of Cape Town University in South Africa. 120 pipes were found, 5 minable, in the Ekati area. Full operation began in October 1998, and Canada became a major player in the diamond game. At Diavik, 41 kimberlite pipes were found, 13 of which contained diamonds, and 4 were rich.
The indicators of diamond are minerals that are normally quite rare at the suface, including garnet, chromite, ilmenite, clinopyroxene, olivine and zircon. The last is the most persistent in sands, and is an excellent indicator that there are kimberlite pipes upstream. Most of these minerals are deeply colored, like red garnet, black chromite, and green clinopyroxene and olivine, and so are easily picked out. They contrast greatly with the usual silica, mica and feldspar. Before the recent entry of Canada, 92% of all diamonds came from Australia, Russia, Botswana, Congo and South Africa.
Brazil also has several diamond regions, but has not made as great an impact on diamond production. Curiously, diamonds have recently (1999) been found in French Guiana, not in kimberlite pipes, but in a greenstone schist, in a mineral called komatiite, at longitude 53°W, latitude 4°N. The Dachine greenstone belt is the remains of the roots of a Proterozoic island arc, where there was once subduction, and the schists were made by the metamorphosis of volcanic material in the Amazonian orogeny (perhaps when South America collided with North America in the Ordovician). This rock, now exposed by erosion, contains only small diamonds. It is interesting that at the time, Africa was in close contact, and this area was opposite the areas of C&ocaret;te d'Ivoire and neighboring countries where alluvial diamonds are now found.
Graphite
The name graphite comes from the Greek grapho, "I write" by allusion to its use in pencils. Graphite indeed leaves a black mark, but it is too soft unless mixed with clay, pressed into thin rods and consolidated by heat. These are called "leads" because soft lead leaves a similar (but not so good) dark mark. The word plumbago for graphite alludes specifically to lead. In ancient times it was probably regarded as yet another form of lead, a more concentrated plumbum nigrum. Graphite pencils do not seem to have been used at all commonly in the ancient world. A stylus on hard wax, or ink on papyrus, were sufficient for most purposes. A specimen of graphite is shown in the figure (© Amethyst Galleries).
Graphite is an allotropic form of carbon, as different from diamond as could be imagined, except for one thing. It, too, is formed by covalent bonding of carbon atoms, and is just as resistant to chemical attack as diamond, and laughs equally well at high temperatures. Graphite does not melt, but sublimes at about 3500°C. Crystalline graphite comes in two forms, α-graphite, which is hexagonal in symmetry, and β-graphite, which is rhombohedral. Most of the time when carbon is reduced to an element, it associates as some form of graphite, though usually not as obviously crystalline graphite. These other forms are often called amorphous graphite, which is good enough for engineers, but all such material is crystalline on a small scale. Graphite is the reason it is so hard to make diamond. Carbon would much rather be graphite under normal conditions. It is more stable than diamond, but diamond never alters to graphite.
 The structure of &alpha-graphite is shown at the right. Pauling gives a free-fusion stereogram of the structure. A carbon atom is located at each vertex of a hexagon, and each line joining two vertices represents a single bond with two electrons. These bonds are 0.142 nm long, intermediate between the 0.154 nm of a single bond and 0.133 nm of a double bond. Each circle represents two delocalized electrons that distribute themselves evenly around the hexagon. To each hexagon corresponds two (6/3) carbon atoms, 6 (12/2) electrons in localized bonds, and 2 delocalized electrons, which accounts for the four valence electrons of each carbon. The delocalized electrons are responsible for the great stability of graphite. Each sheet of hexagons is a separate molecule, a natural colloid that is all surface. The sheets are held together by van der Waals forces that are much weaker than the covalent bonds. The fluctuating charges in one sheet induce opposite charges in the other sheet, so the sheets attract. In the α-graphite, they are 0.340 nm apart. Heat and electricity flow easily in a sheet, with difficulty normal to the sheets. The resistivity normal to the sheets is 10,000 times the resistivity in a sheet. A sheet can stick to another plane surface, as well as adsorb molecules and exhibit all the usual surface phenomena. The uses of graphite as a lubricant and as an adsorbent depend on its surface.
The structure of &alpha-graphite is shown at the right. Pauling gives a free-fusion stereogram of the structure. A carbon atom is located at each vertex of a hexagon, and each line joining two vertices represents a single bond with two electrons. These bonds are 0.142 nm long, intermediate between the 0.154 nm of a single bond and 0.133 nm of a double bond. Each circle represents two delocalized electrons that distribute themselves evenly around the hexagon. To each hexagon corresponds two (6/3) carbon atoms, 6 (12/2) electrons in localized bonds, and 2 delocalized electrons, which accounts for the four valence electrons of each carbon. The delocalized electrons are responsible for the great stability of graphite. Each sheet of hexagons is a separate molecule, a natural colloid that is all surface. The sheets are held together by van der Waals forces that are much weaker than the covalent bonds. The fluctuating charges in one sheet induce opposite charges in the other sheet, so the sheets attract. In the α-graphite, they are 0.340 nm apart. Heat and electricity flow easily in a sheet, with difficulty normal to the sheets. The resistivity normal to the sheets is 10,000 times the resistivity in a sheet. A sheet can stick to another plane surface, as well as adsorb molecules and exhibit all the usual surface phenomena. The uses of graphite as a lubricant and as an adsorbent depend on its surface.
The single bonds in graphite are formed by sp<2 hybrid orbitals that lie in a plane at angles of 120°. The remaining electrons are in a pz orbital, half above the plane, half below. These orbitals overlap to form the circular orbitals above and below the plane that accommodate the delocalized electrons to give graphite considerable "resonance" energy.
Although diamond is a semiconductor, graphite is classed as a semimetal. Its valence band is filled, and its conduction band is empty, at absolute zero, but the energy gap Eg = 0. Therefore, there are always mobile electrons in its conduction band, and mobile holes in its valence band, at nonzero temperature. These charge carriers are not responsible for its bonding, as in a metal. They are, however, responsible for the semimetallic lustre of graphite. The black of graphite is due to its fine division, usually colloidal, which absorbs light effectively.
One way to make a colloidal graphite is to burn things that make a smoky flame, like resins, and to catch the soot on a cool surface. The soot is called lampblack. In Latin, this material was called fuligo, fuliginis, and was used to make black paints and the important substance atramentum librarium, or ink. To make ink, you ground fuliginem in a mortar with some gum, gummi, perhaps from the acacia, and a little water. The colloidal graphite was then peptized by the gum, forming a stable sol that was the ink. This recipe was discovered in Egypt when writing on paper was invented, and is now known as India ink. The formula is given in Vitruvius. Ink, paper and writing all came to the west from Egypt. Ink was also discovered in China, where gelatin was used instead of gum. Writing in a different fashion also came from China, with paper made from textile fibres. This excellent ink is still used today in drafting, a survival of a very ancient technique.
Graphite is found in nature in three forms, known in the trade as crystalline flake, crystalline lump, and amorphous lump. Crystalline flake graphite occurs as flakes in the metamorphic rock mica schist. The world's best deposit is in Madagascar, though it is widespread. In Madagascar, 13 tons of schist yield 1 ton of 90% graphite flake. This is some of the most desirable graphite in trade, called "crucible grade" and also used in lubricants, where all abrasive impurities must be absent. A primary use of graphite is in making refractory crucibles and other objects that must stand extreme heat. Graphite vanes were used to steer the V-2 rockets in world war II.
Crystalline lump occurs in veins associated with intrusive pegmatites in Ceylon. The graphite veins radiate from the pegmatite inclusion. The source of the graphite is probably the reduction of the common volcanic gas carbon dioxide. Amorphous lump graphite is produced by the alteration of coal by nearby granite intrusions, which cooks out the volatile ingredients in a kind of natural coke oven. In fact, there is a continuous progression through anthracite coal to graphite. The structure of coal is different from that of graphite, however. These graphites can be used to add carbon to steel, and other uses that do not require high purity. The largest veins of amorphous graphite are in Sonora, Mexico, bedded in a triassic sandstone. These were indubitably coal deposits cooked by nearby intrusions. Similar deposits occur in Korea, which supplies most of the world's lump graphite. New Mexico and Rhode Island also have vein graphite deposits.
Dr Acheson found how to make good graphite in an electric furnace. It seems to be competitive with natural graphite, and is generally purer. Graphite can be peptized by tannin to make an aqueous sol called aquadag, and a dispersion in oil called oildag. The "dag" syllable seems to be a term for deflocculated graphite, a product of the same company. Aquadag can be used to deposit a conducting graphite layer on the interior of a cathode-ray tube to complete the electron circuit. Graphite is used for the cathode in ammonium-zinc and alkaline electrical cells because of its inertness and conductivity, and as carbon arc electrodes because of its high sublimation temperature.
Graphite is a good lubricant when in fine flakes, even when dry. This is not because the graphite layers slide easily on each other, because they do not, but stick persistently. When a layer has adsorbed neutral molecules on both sides, however, it does become slippery. It tends to plate metal surfaces, so it does not rub off.
Carbon black is the colloidal graphite used in rubber products, and is identical to lampblack. It is usually made by burning methane in limited air. Rubber is very susceptible to abrasive wear, and tires would last a very short time unless the rubber were mixed with large quantities of carbon black, which gives it wear resistance. A chemical reaction is probably involved, part of the vulcanization process. The general black color of rubber products is evidence of the use of carbon black. Pure rubber is a pasty whitish color.
Thomas Edison found that the resistance of carbon granules depended on the pressure to which they were subjected. By connecting an acoustic diaphragm mechanically to a button of carbon granules, the resistance could be made to vary in accordance to the acoustic pressure, so that when a current was passed through the button an amplified replica of the acoustic signal was produced. This gave a much stronger signal than the Bell type of metallic diaphragm, permanent-magnet microphone, and made long-distance telephony possible. Edison, as is well-known, also used carbon filaments in the first incandescent lamps. The search for the best material to carbonize is a fascinating story. Bamboo fibres turned out to be the best.
Carbon paper is another useful invention largely ignored today (though carbon paper can still be purchased). It uses a film of carbon black on thin paper for the purpose of making multiple copies simultaneously by being struck with a type head of a typewriter, or embossed with a stylus held in the hand. An early use was making multiple copies all at one time of telegraphic railway train orders, messages that had to be precisely correct, so they were guaranteed to be identical. In the business world, a typist could make a file copy at the same time as the original letter, or make a copy for another recipient. There had been ways of making multiple copies before carbon paper, but carbon paper was far superior to them.
 Black powder is black because it contains graphite from charcoal. Gunpowder is made from potassium nitrate, charcoal and sulphur very intimately mixed by grinding when wet. This is a large subject that cannot be adequately treated here, except to say that making gunpowder is somewhat more difficult than would be guessed, and there are many tricks involved. Gunpowder actually burns very rapidly rather than exploding, and this is what is desired in a firearm. Gunpowder Creek in Wyoming was so-named by the Cheyennes because of the coal that crops out on its banks. Gunpowder was the only black powder that they knew.
Black powder is black because it contains graphite from charcoal. Gunpowder is made from potassium nitrate, charcoal and sulphur very intimately mixed by grinding when wet. This is a large subject that cannot be adequately treated here, except to say that making gunpowder is somewhat more difficult than would be guessed, and there are many tricks involved. Gunpowder actually burns very rapidly rather than exploding, and this is what is desired in a firearm. Gunpowder Creek in Wyoming was so-named by the Cheyennes because of the coal that crops out on its banks. Gunpowder was the only black powder that they knew.
Oxides
Carbon forms three oxides: carbon monoxide, CO; the dioxide, CO2; and the unimportant but curious carbon suboxide, C3O2. The suboxide is a colorless gas condensing to a liquid at 7°C. In water, it hydrolyzes to malonic acid, HOOC-CH-COOH, of which it is the anhydride. Its structure is linear, O=C=C=C=O.
The Lewis structures of CO and CO2 are shown at the right. Each line represents two electrons in a covalent bond; assign one electron to each atom joined by the bond. The dots represent electrons belonging to a particular atom, here the oxygens. Let the oxygen have a formal charge of +6, the carbon +4, each electron -1. Then the formal charges are as shown. The center structure is nonpolar, while the outside structures are polar in opposite directions. The state for the molecule as a whole is a linear combination of the three structures. The ability to choose the relative amounts of the structures allows the formation of a state of lowest energy. The is rather inappropriately called "resonance" in chemistry. There is no resonance involved, just the freedom to adjust the relative amounts. The amounts of the two polar structures are equal, so the molecule is nonpolar overall. The resultant lengths of the bonds are shown at the right. The possibility of "resonance" makes these molecules especially stable.
Carbon monoxide is a very stable diatomic molecule. It is a colorless, odorless gas at STP. Its molecular weight of 28 makes it just a tad lighter than air. It burns to carbon dioxide in the presence of oxygen, releasing a good deal of heat (67,700 cal/mole), and reduces iron oxide to the metal, becoming carbon dioxide. Banefully, it combines tightly with hemoglobin in the blood, taking it out of action and suffocating from within. For this reason, it is a deadly poison in air that is breathed. One part in 100 is rapidly and irreversibly fatal. One part in 500 brings unconsciousness in 30 minutes, death in three hours. Otherwise, it is just fine. Carbon monoxide is created when carbon is burned in a deficiency of oxygen, as in charcoal fires, internal combustion engines, poorly drafted and vented heaters and furnaces, fuel gases, and in methane explosions in coal mines. The symptoms of inhalation of small quantities are headache and nausea. It is treated by inhalation of oxygen containing 5% carbon dioxide. Carbon monoxide can be detected by the blackening of paper soaked in palladous chloride solution, or in smaller quantities by electronic sensors.
CO is produced endogeneously in the body in the breakdown of the heme molecule when an erythrocytes' 120-day life is over. If it were not for a molecular adaptation in myoglobin and hemoglobin, the proteins that carry oxygen in the muscles and blood, this would make enough CO to block the globins fatally. As it is, about 1% of the globin is blocked by CO in normal circumstances. A free heme is 25,000 times as sensitive to CO as it is to O2, but the globins only 200 times more sensitive. Life is complicated.
Carbon monoxide can be removed from internal combustion engine exhaust by adjustment of the engine, but this increases the NOx emissions, which are even more poisonous. No hay lonche gratuito. Catalytic converters mainly catch unburned hydrocarbon fragments, and don't do much with CO and NOx.
Carbon monoxide can be catalytically hydrogenated to make methanol (wood alcohol), which is a violent poison when taken internally, but nice enough otherwise, and a good solvent. With chlorine, it forms carbonyl chloride, COCl2, also known as phosgene, a rather unsuccessful war gas, but a useful chemical intermediary in making dyes. The patented compound Hopcalite (Johns HOPkins and U. of CALifornia) oxidizes CO to CO2 in gas masks. It is a mixture of manganese dioxide with copper and cobalt oxides, and must not be exposed to the slightest amount of moisture.
In coal mining, CO is called black damp, and occurred after there had been an underground fire or explosion. Canaries were used to detect it, since the birds were more sensitive than the miners to its effects. The safety lamps of Davy and Stephenson were used to detect the presence of methane, or fire damp, (by an enhanced flame) without setting it off. CO also had an effect on the flame. CO2 was called choke damp, because it suffocated. The word "damp" here is cognate to German "dampf" and simply means a gas.
Carbon monoxide forms complexes called carbonyls with metals, like nickel tetracarbonyl, Ni(CO)4. The carbons are tetrahedrally bound to the nickel, with the oxygens on the outside. There is "resonance" between the uncharged structure and structures with various charges on the carbon and oxygen that bind the molecule. Without "resonance" there would be no binding at all. Nickel carbonyl is a volatile liquid melting at -25°C and boiling at 43°C. It is formed by passing CO over warm Ni powder, then it is condensed and distilled for purity. The pure carbonyl is then pyrolytically decomposed at 150°C to deposit pure nickel. Carbonyls are usually inflammable clear liquids easily decomposed by heat.
Carbon dioxide is a very stable linear triatomic molecule, generally represented by O=C=O. Its molecular weight of 44 shows that it is heavier than air. It is colorless, odorless and nonpoisonous. It does stimulate breathing, however. The critical temperature is 31.1°C, so it is easily liquefied by pressure alone, and is supplied in steel tanks. The liquid freezes under 5.1 atm pressure at -56.6°C to a white solid, dry ice. This is the triple point at which gaseous, liquid and solid can coexist. At 760 mm CO2 pressure, dry ice sublimates at -78.5°C, and can freeze ice cream harder than a rock. At lower pressures, the sublimation temperature is even lower. It provides pure cold, without messy liquids. It is a normal minor constituent of the atmosphere, about 350 ppm by volume. Nevertheless, this small amount supplies all the food in the world for all living things, and is part of a complex natural cycle. Carbon dioxide, incidentally, dissolves in rubber and diffuses through rubber faster than helium or hydrogen does.
Carbon dioxide is quite soluble in water. At 1 atm, 1 litre of water dissoves 1.713 litres of dioxide, making a pleasingly acidic, effervescent solution. The equilibrium concentration in the solution is proportional to the carbon dioxide pressure. Soda water is charged with carbon dioxide at 4 to 5 atm pressure (50-65 psi), which slowly evaporates if the solution is open to the air, more rapidly if the solution is agitated. Supersaturation is easily observed when an opened can of soda is poured into a glass, and gas is then copiously evolved. This supersaturation is very desirable in a beverage. All rainfall is slightly acidic, even when absolutely pristine. Water in equilibrium with the usual atmospheric content of CO2 has pH 5.7. The carbonate ion is formed by the action of the water: CO2 + H2O ↔ H2CO3 ↔ H+ + HCO3- ↔ 2H+ + CO3--. This series of equilibria show that the solution contains a number of molecular constituents. The ionization of water means that the hydroxyl ion concentration is very low. The carbonate ion has a threefold axis of symmetry, and is very stable, forming a wide variety of important salts. H2CO3 is called carbonic acid. Its true dissociation constant is 4.4 x 10-4, so it is a fairly strong acid. However, the concentration of H2CO2 is so low that the actual H+ concentration is also small. The apparent dissociation constant [H+][HCO3-]/[CO2] is only 4.2 x 10-7, so it is effectively a weak acid. Although real enough in solution, it cannot be isolated out of water as a crystalline substance, and always exists in equilibrium with dissolved CO2. Carbonic acid seems to speed alcohol from the stomach to the blood stream, an interesting effect of carbonated drinks. The dissociation constant of bicarbonate to carbonate is 4.8 x 10-11, and of water 1 x 10-14. At a carbon dioxide pressure of 2.9 atm, the pH of the water solution is 3.4; at higher pressures it does not go much below 3.3.
Calcium carbonate is a very important carbonate, found in nature in limestones. Its rhombohedral crystals are really just carbonate ions, all pointing in the same direction, made electrically neutral by Ca++ ions at strategic locations. Clear crystals are called calcite. There is also an orthorhombic variety, called aragonite. It is quite insoluble, but calcium bicarbonate, Ca(HCO3)2, is not. When water that has absorbed some carbon dioxide trickles across it, the bicarbonate ions pick up some calcium from the carbonate, and wash it away in solution. This is an important weathering process that explains the weathering of limestones in a moist region along cracks and passages, producing great and small caves, underground streams, and all the wonder of Karst topography. (Karst is a limestone area in the Balkans that is an example of this.) This is part of a subcycle of the great carbon cycle in which animals in the sea make shells of calcium carbonate, which becomes limestone, which is raised above sea level, which is rained upon, and which returns to the sea as bicarbonate. Water containing calcium bicarbonate is the classic "temporary" hard water, since boiling deposits the carbonate as scale. The calcium forms the familiar insoluble scums with soap, so "softening" water means removing Ca++ ions, and Mg, Fe and Mn ions as well. "Permanent" hard water contains anions such as SO4-- that will not precipitate the cations. In this case, the addition of carbonate is necessary, perhaps as washing soda, Na2CO3.
Carbon dioxide in water also weathers silicates to calcium bicarbonate and silicic acid, both of which are soluble. Carbon dioxide is released when silica and calcium carbonate react in boiling water to make calcium silicate and carbon dioxide. This is the origin of the "geyserite" produced around hot springs and geysers, and is the reverse of the first reaction. The weathering of feldspars to kaolin is one way carbon dioxide is removed from the atmosphere. Another is the fixing of carbon dioxide by organisms to calcium carbonate. Photosynthesis also removes carbon dioxide in large amounts, while certain bacteria use carbon dioxide in chemosynthesis. Most photosynthesis occurs in the surface layers of the ocean, it should be remembered, not by the more visible green land plants. The ocean contains 20 to 30 times as much carbon dioxide as the atmosphere, and is dominant in the carbon dioxide cycle. All this is very difficult to estimate quantitatively. It is no wonder that computer programs may seem to give observed variations; such programs are constrained to do so.
Carbon dioxide can be made by "burning" limestone. With heat, limestone decomposes according to CaCO3 → CaO + CO2. This is a classic way of making the dioxide. CO2 is a by-product of lime burning, cement making and brewing. Lime, CaO, is "slaked" in water to form the hydroxide: CaO + H2O → Ca(OH)2. Mixed with sand and water, the hydroxide forms mortar. The hydroxide first crystallizes rapidly, and the morter sets. Then it gradually reacts with the carbon dioxide in the air to return to the carbonate, and hardens even more. Carbon dioxide is also made in fermentation, when enzymes catalyze the reaction turning sugar into ethyl alcohol and carbon dioxide. This is the major source of industrial CO2. Yeast contains the enzyme zymase to accomplish this creditable task. Unlike methyl alcohol, ethyl alcohol is a good drink that is the source of much government revenue and a boon to the human race. Beer is an exceptionally healthy beverage that we should consume more of.
The carbon dioxide industry has been well developed. It supplies liquid carbon dioxide in strong steel cylinders, and solid carbon dioxide in compressed bricks. The liquefaction of gases was introduced by Faraday, while Thilorier discovered solid CO2 in 1835. Solid CO2 can be made by direct free expansion of liquid CO2 to form CO2 snow, which is then compressed in bricks. If no other source of the gas is available, it can be made by burning coke in air. The flue gas is scrubbed over limestone chips to remove SO2 as calcium bisulfite, then the CO2 is absorbed by an alkaline solution (Na2CO3, for example, called "lye") in a coke-filled tower and released in purified form by boiling the lye. The gas is then dried and compressed, and condensed to a liquid, eliminating the nitrogen. Some sources of gas, such as fermentation, may introduce unwanted odors and impurities. Cylinders holding 50 or 20 lbs of the liquid, or smaller cylinders for certain applications, are filled to about 60%. If they are overfilled, expansion of the liquid can produce dangerous stresses.
Liquid carbon dioxide has had many uses. As a compact source of power, carbon dioxide can be used in place of compressed air. Before electricity was widely available, it was used to operate some railway signals in isolated locations. Similarly, it was used to operate bell buoys for navigation. It was used in coal mining as an explosive ("Cardox"), by heating it in a closed cylinder until a shearing disc was ruptured and the high pressure (10,000 - 30,000 psi) released fractured the coal. It was used as a safe refrigerant until displaced by Freons. Now that Freons are out of favor, it may again be used for this purpose. The greatest use of liquid carbon dioxide is in the carbonation of beverages, and in raising beer. It not only can provide the power to lift beer from the cellar to the tap, but preserves the carbonation in doing so. Dry ice is used where other methods of refrigeration would be heavy or inconvenient, particularly in the distribution of ice cream. It provides an inert atmosphere for processes at moderate temperatures (at high temperatures, it dissociates to CO + O2, so cannot be used for welding).
Carbon dioxide is found naturally as a major consitituent of volcanic gases, and in caves, mines and wells. At Price, UT, a well produced 98% CO2 at 760 psi. Some oil wells in western Colorado produced an emulsion of CO2 and crude oil, and were called "ice cream wells." It occurs in many naturally carbonated spring waters, as at Saratoga Springs, NY (in equilibrium with 21 psi CO2), as well as at famous locations such as Seltzer, Spa, Vichy and Apollinaris. The CO2 in these waters helps them to dissolve minerals, hence they are "mineral" waters. At the Grotta del Cane near Naples, the gas is 70% CO2, 24% N2 and 6% O2, and is definitely fatal. The Stygian Caves in Yellowstone, and the Lachersee in Germany, also are associated with carbon dioxide. Fatal properties of these gases are associated more with the lack of oxygen than the presence of carbon dioxide. Compositions of 88% N2, 10.6% CO2 and 1.4% O2 (in a manhole) and 48.5% N2, 38% CO2 and 13.5% O2 (in a silo filled with green corn) have proved fatal. On the other hand, a mixture of 5% CO2 and 95% O2, called "Carbogen," is used to stimulate breathing in CO poisoning and in termination of anaesthetics. The greatest stimulation of breathing occurs at about 7%, but concentrations over 25% or 30% tend to terminate breathing. Carbon dioxide plays a very important role in regulating breathing in normal concentrations. A human exhales about 900g of CO2 per day. Since there are about 6 x 109 humans at present, this is not a negligible contribution to greenhouse gases.
Sodium carbonate, Na2CO3, and sodium bicarbonate, NaHCO3, form clear hydrated crystals, but are more commonly known as white powders. They are the household alkalis, as vinegar is the household acid. Sodium carbonate decahydrate is called washing soda, since it is used to precipitate calcium carbonate to soften water for laundry, and sodium bicarbonate is called baking soda, since it is a source of CO2 to leaven baked goods. When dissolved in water, the carbonate ion ties up all the H+ ion in scarcely-ionized molecules, leaving the OH- to rule. Sodium carbonate produces a strongly alkaline solution that irritates the skin and is very unpleasant to drink, but bicarbonate is an excellent remedy for excess stomach acid. A half-teaspoon in 5 ounces of water will cure indigestion at once with a satisfying belch, at little cost. This is the reason more expensive medications are touted, and bicarbonate ignored.
Sodium carbonate and bicarbonate are found naturally as the mineral trona, but much is produced artificially, since natural deposits are uncommon and often impure. It is found in evaporites in arid regions, and in fossil evaporites underground. The LeBlanc and Solvay processes have been used to manufacture the carbonate, both of which begin with salt, and the carbonate supplied by limestone. Pure sodium carbonate is a prime ingredient of soda glass. It is also used in making paper. Boiled with slaked lime, and then with fats, it makes soap. Soap dissociates in water to a molecule that has a polar end, and a nonpolar fatty end. It peptizes oil to make micelles with the polar ends on the surface that disperse in water and can be rinsed away.
Baked goods are foams of CO2 in a gel of glutin, making grains easily digestible and pleasant. The CO2 can be provided by yeast, which not only ferments the sugar, but makes its own sugar from the starch in the flour. It is usual to add some sugar to make the job easier. The alcohol evaporates in the heat, incidentally. The gas can also be provided by sodium bicarbonate, and then we have soda bread in place of yeast bread. The American "biscuit" and the Scottish "scone" are soda bread. American "corn bread" is soda bread carrying corn meal, which lacks the ingredient necessary to make a foam, but provides sweetness. Without the strong wheat flour, it would be just ghastly "pone." Soda bread can be just as delicious as yeast bread, and is less trouble. The only thing missing is the yeasty flavor. Two reactions are generally used to produce the carbon dioxide. The first is with lactic acid, which is present in sour milk, buttermilk, and milk "soured" on the spot with a little vinegar. The results are sodium lactate and gas. Another, using dry ingredients that activate only when moistened, is with potassium hydrogen tartrate. The dry mixture is called baking powder. The hydrogen is exchanged for the sodium, and we have sodium potassium tartrate, or Rochelle salt, and gas. The gas results from the dissociation of the carbonic acid, as usual. The reactions are illustrated with structural formulas in the diagram. In each case, a hydrogen and a sodium change places. These are very familiar and useful reactions that every cook should understand.
The old red and brass soda-acid fire extinguishers that used to hang in school corridors had a tank full of sodium bicarbonate solution, and a bottle of sulphuric acid with a loose stopper suspended at the top. When taken down and inverted, copious carbon dioxide was evolved, which pressurized the closed top (which was now the bottom) and squirted water out of the nozzle to be directed at the fire. The CO2 helped to blanket the fire as well, but inefficiently. These were cheap and effective, but being cheap, were eliminated in favor of more advanced and expensive pressurized extinguishers that leak propellant and are duds when an emergency finally arises. Schools don't have too many oil fires, but when one does happen, a water extinguisher does not work. Foam extinguishers are much better, especially when chemically propelled in the old fashion and not by pressurized nitrogen.
As carbon dioxide is produced by fermentation, it is also produced in the formation of coal and petroleum from organic matter, probably with the aid of bacteria. This carbon dioxide usually escapes, but is occasionally found where it has been trapped. In Wyoming, Colorado and New Mexico, there are many carbon dioxide wells that produce the gas from deep porous rocks. This is probably gas that has never been in the atmosphere. It is used for pressurizing natural gas reservoirs, and in secondary recovery of petroleum, where it also has a solvent effect.
Carbon dioxide is called a "greenhouse gas" because it absorbs and emits infrared radiation. This slows the radiation of heat by the atmosphere into space in the same way that the glass windows of a greenhouse do. The visible light with its energy can get in, but the infrared radiated by the things it heats inside cannot get out as easily through the glass, so the greenhouse stays warm in the winter without the expense of fuel. The more carbon dioxide, the larger the effect. Carbon dioxide is not the only greenhouse gas, and is actually a minor one. Water is the most important greenhouse gas. Methane and nitrous oxide are more effective than carbon dioxide, but present in smaller amounts. If it were not for the greenhouse effect, the surface of the earth would be as cold and inhospitable as the surface of Mars (almost). Stick your hand out the window the next time you are flying at 35,000 feet to see what the temperature is like out there. It is cold enough to freeze the spherical objects off a copper-alloy primate. Long-range climatology is a delicate balance that we do not know enough about to say anything for certain. There must either be global warming or global cooling (the only choices) and we seem to be stuck with the warming. It has been much warmer, and much cooler, in the past, mainly warmer. Causes we do not understand. What we can do is agitate for more research support.
Fossil Fuels
What are called fossil fuels are naturally-occurring substances composed mainly of carbon and hydrogen. On combining with the oxygen of the air, they yield water, carbon dioxide and, most importantly, large amounts of heat. They may be solids, liquids or gases. All are found in the upper crust of the earth, but only in sedimentary rocks that have not been subjected to great heat or pressure. All are the products of former life, produced by photosynthesis from water and atmospheric carbon dioxide, with the input of radiant solar energy. Their supply is strictly limited, and compared with the current demand, small. This applies with special emphasis to the fossil fuels available at low cost.
Fossil fuels appear in three major forms. Carbonaceous deposits are mainly carbon, with very little hydrogen and other elements. These are represented by coal. Bituminous deposits include hydrogen as well as carbon, and are represented by natural gas (gaseous), crude oil (liquid) and bitumen, asphalt or tar (solid). They are called, collectively, petroleum, though this term is popularly applied specifically to crude oil. The third form is pyrobituminous deposits, which must be distilled before they can be used. This form includes oil shales, and special varieties of coal. All fossil fuels are the remains of living matter. Coals arise from forest plants the size of trees, whose structures are clearly recognized in the coal. No coal is earlier than Carboniferous, since there were no appropriate plants before then. The other fossil fuels are the remains of microscopic organisms, chiefly diatoms and similar sea plants, which have existed from the Ordovician to the present in very large numbers. This is shown by the existence of optically active substances in them, and the presence of porphyrins. All fossil fuels have been produced with the aid of anaerobic bacteria that extract oxygen and evolve methane. There is no possiblity of an inorganic origin of fossil fuels.
Most terrestrial carbon is in the form of carbonates in sedimentary rocks, or as dissolved carbon dioxide in the seas, about 4.6 x 1015 tons. Some 7.1 x 1012 tons is in the form of coal and similar substances, while 1.1 x 1012 tons is in soil and living matter. The amount of carbon in petroleum is negligible. Of the oxygen that is not in water, about 8.0 x 1015 tons is in sedimentary rocks, 1.2 x 1015 tons is in the atmosphere, but only 1.2 x 1013 tons is dissolved in the sea. Note that carbon dioxide is very soluble in water, while oxygen is not. These figures are from Holmes (1945). Originally, the carbon dioxide was all in the atmosphere, while the oxygen was all in water. Life processes, mainy photosynthesis, have been responsible for the current state of affairs.
Plants on land use sunlight to take the hydrogens from water to make hydrocarbons from atmospheric carbon dioxide, releasing the oxygen as a waste product. This is done by the active membranes in chloroplasts, which use chlorophyll to absorb light at long and short wavelengths to gather the necessary energy. Plants still respire as all modern life does, by oxidizing hydrocarbons in mitochondria which take in oxygen and release carbon dioxide. Photosynthesis and respiration are complementary processes. The organisms that originated these ingenious mechanisms were incorporated by symbiosis in the cells that protected them from the change in the atmosphere from a reducing, carbon dioxide-rich condition to an oxidizing, oxygen-rich condition. There is no evidence of the first life, except for these relics that are absolutely essential cellular organelles. The palaeozoic carbonates are the very last stage in this process, most of which happened long before.
Coal
Towards the end of the palaeozoic, a new way of sequestering atmospheric carbon occurred. The new land plants of the Carboniferous manufactured huge amounts of carbohydrates that were largely structural elements, not food, except for the bacteria who knew how to digest cellulose and live in the stomachs of plant-eaters. Cellulose is all energy-rich sugar, but locked up so devillishly that it is highly insoluble and very durable. This cellulose was buried in bogs away from the air that aerobic decay bacteria required for their functioning, and only those microorganisms that fermented remained, slowy abstracting the hydrogen and oxygen from the buried plant material, turning it into a colloidal jelly of what remained of the cellulose, which by now was something quite different. Deeper burial slowly squeezed out the water, and all bacterial action ceased, leaving only a complex black material of very complex structure, and consisting mainly of carbon. This material is coal.
Coal is produced only under special conditions of deposition, when the plant growth can flourish, followed by shallow swamps, followed by burial in sediments. The late Carboniferous (Pennsylvanian) was the first such time, when the conditions of sedimentation were cyclic, creating repetitive cyclothems of clay, coal and shale, in which the remains of the forests of Lepidodendron and Sigillaria, large fern-like plants, were preserved. Normally, aerobic decay processes would eliminate all trace of the former plants, returning their carbon to the atmosphere. The late Cretaceous was another such time. Most U.S. coals are either Pennsylvanian or Cretaceous. In other parts of the world, Triassic and Jurassic coals are found. Coal does not seem to be forming at the present time, although there are peat bogs, but no assurance that these bogs will eventually be coal. They could just dry up and disappear by decay.
A typical coal seam begins with the seat earth at the bottom, the original clay bottom of the swamp in which the forests grew. Then there is the coal, from numberless generations of strange trees, but no fossils of fish, showing that the swamp was black and lacked dissolved oxygen to support life. When trunks fell into the bog, they did not decay, but were attacked by the anaerobic bacteria that turned them first into a jelly, and then to peat, as the oxygen was used up and the hydrogen spent. By pressure, the peat was dried and by further bacterial action was altered to coal. Above the coal is fossiliferous sandy shale, or sandstone, showing that the bog had been submerged, the forest drowned and covered with sediment and fresh water. The upper part of the coal often contains evidence of the fallen trunks from the last generation. There may be a layer of iron ore, the result of more bacterial action when the forest was gone, and before freshwater conditions returned. This sequence is repeated over and over in the Coal Measures, as these rocks are known, and the seams extend over considerable areas.
Coal is a valuable fuel resource because it is elemental carbon, which can be burnt in the atmospheric oxygen to release heat. This, of course, only reverses the process that formed it, where the heat input was from the sun. Coal is just solar energy, released from where it has been stored for millions of years. There is only a finite amount of coal, and the sea and atmosphere can probably accommodate all of its carbon as carbon dioxide, if it were released at a reasonable rate. The problem is, however, is that humans tend to spend their money all at once, and burn all their coal the same way, which may cause a temporary disturbance. The carbon dioxide could be used to make more cellulose, again sequestering the carbon and releasing oxygen, while creating a new store of fuel. But humans will be long gone before this slow process can be realized.
Petroleum
A much smaller amount of carbon is sequestered as petroleum than as coal. Petroleum is to carbon as silica is to silicon. Silicon is not found in elemental form in nature, because it would much rather exist as silica, which has a much lower free energy. Carbon will combine with oxygen as well, to form the gas carbon dioxide and the carbonates derived from it. However, carbon will also combine with hydrogen to achieve a state of lower free energy than it can by forming graphite (and certainly diamond). Instead of the giant molecules of silica, we get the modest molecules of the hydrocarbons. The original source of the carbon must be living tissues, from which something must abstract the oxygen. When this is done, we have petroleum, which can be a gas, a mobile liquid, or a solid.
This seems to have happened when the black muds into which dead diatoms and other algae had fallen and had been preserved, began to change into shales. If this occurs in the absence of air, we have something similar to the conditions for the formation of coal, but the great masses of cellulose are missing, and we have the remains of bacteria, algae, and other simple forms of life. Microorganisms again go to work, removing the oxygen. As they do so, gases may be evolved, such as methane, CH4. The same thing occurs when coal is formed, incidentally. In some coal beds, the methane has escaped, but in others its departure has been hindered, and the coal is called gassy. This gas is now being exploited in the United States where it exists in quantity. It will quickly be exhausted, but represents a windfall.
The liquids may escape from this shale, under the weight of overlying sediments, gas pressure, or floating on salt water, if there are permeable rocks adjoining. The carbon remaining turns the shales black, showing that they once held life, and that they were buried in anaerobic conditions. Most shales are not black, and whatever life was in them decayed and returned to the winds and waters before the rock was formed. In other cases, the black shales are clearly the source rocks for the liquid and gaseous petroleum that may be trapped nearby. Petroleum is much simpler structurally than coal. Of course, the original form of the carbon has no influence, since the petroleum has moved from the place it was formed. A liquid petroleum, crude oil, may be largely straight-chain hydrocarbons, or paraffinic, or contain 6-membered rings and double bonds, and be asphaltic. Underground, it may have smaller molecules dissolved in it that come out in bubbles when the pressure is reduced.
Petroleum is a double-barreled energy reserve, because both the carbon and the hydrogen can be burned to create heat. It is still mainly burning carbon, however. Natural gas, represented by CH4, is 75% C and 25% H. Gasoline, represented by hexane, C6H14, is 84% C and 16% H. Liquid petroleum fuels are specially attractive because they are easy to handle and have a large energy density. It is much easier to carry around gasoline than the equivalent in storage batteries. Natural gas was easy to handle, but the energy density was so low that it was not economical to transport, and much was wasted, often flared off uselessly (but economically). The low price of fossil fuels makes such waste common. Using natural gas to fire boilers is a tragic waste of a valuable resource, but it is even encouraged. Petroleum is much more valuable as a chemical feedstock than as a fuel, which the world will discover to its sorrow in a century or so. Coal is the only fossil carbon whose fuel use can be justified.
Petroleum exists in several forms in the earth. Liquid and gaseous petroleum are the most known, because they are easily gathered. Petroleum-saturated tar sands exist in northwestern Canada. Huge amounts of gray-green shale are permeated by a waxy petroleum called kerogen in western Colorado and Utah. In fact, illuminating oil was first distilled from oil shales in Scotland, before liquid petroleum was widely known. The distinctive thing about tar sands and oil shales is that real work must be done to get at the petroleum, and this work will be expensive and environmentally damaging. We have broken into the bank of stored energy, and are only sweeping up the cash we find lying around in view, not sweating to pick locks and pry open drawers.
Liquid and gaseous petroleum, crude oil and natural gas, lurk under pressure in the pore space of sedimentary rocks. The rocks must not only be porous, but also permeable, so the petroleum can flow. This is usually the case, since the petroleum had to leak in in the first place. However, nature is satisfied if this happens in ten thousand years, and we are a bit less patient. If we drill a hole to a porous, permeable rock holding petroleum, it squeezes into the hole and usually up the hole to the ground. If we want gas, the gas must do this in any case. If crude oil only goes part way up, we can pump out what is in the hole and let the hole fill up again. You cannot suck petroleum out of the rocks. Of course, we can help. Gas can be forced down a hole to give extra pressure. The gas is usually carbon dioxide, which also makes the oil less viscous. Water can be forced in from wells surrounding the oil, to wash it forward. These are really desperation measures, but can extend the life of many oil fields that would have been abandoned in the past. If oil commanded a realistic price (perhaps twice or three times what it now brings) these efforts would be more general, and much oil would be saved that is now going to waste.
Exploration for oil is now a very refined and powerful process, and has rummaged literally everywhere, from pole to pole. However, there is no process that can detect the actual presence of petroleum except the drilling of a hole, and the nature and contents of the reserve cannot be estimated until some oil has actually been produced. It is always dangerous to make predictions of such uncertain things as the presence of oil, but saying that there are no more large, or even medium-sized oil fields left to be discovered in the United States is about on the same level as predicting that a large asteroid will not collide with the earth and wipe out a continent in the next century. Either could happen, but not bloody likely. The decline of domestic production, as a ten-year running average, is inevitable. Venezuela, Mexico, Canada and Nigeria will slowly follow, and eventually even Iran, Iraq and Saudi Arabia. The current demand for oil is huge and growing, the supply finite. It is like a pile of cash, so easy to scoop up as many armfuls as we want, but the pile just gets smaller. As late as 1973, the United States could dominate the world oil market, but now it is powerless. The United States was once extremely rich in cheap petroleum. Now we are scraping the sides of the bowl.
Charcoal, Coke and Carbon Black
Charcoal, coke and carbon black are all colloidal forms of carbon, and this should be kept in mind while considering them. Being colloidal means that they are effectively all surface, though this may not be evident in the bulk.
In Latin, carbo, carbonis was charcoal, and a carbonarius was a tradesman who made it. In German, Kohl was charcoal, and a Köhler was a tradesman who made it. In English, coal was charcoal, and a collier a tradesman who made it. Charcoal, a smokeless fuel, was the preferred fuel for the home, used in a brazier to cook or to heat a room. Charcoal was also the fuel for the smelter of iron and other metals, and for the smiths who worked it at their forges, because it was pure. To make charcoal, hardwood trees were cut into sticks, which were piled in a dome-shaped heap and plastered over with mud and turf. A typical heap might include 15 to 45 cords of wood (a cord is 128 cuft). A hole was left at the top, and an opening at ground level on one side. The fire was lit at the opening, and when things were burning nicely, the opening was nearly blocked up to limit the air supplied to the fire. Dense smoke came out the hole in the top, as the volatiles were burnt to drive the rest out, at a temperature of about 400°C. It could take as long as a fortnight to complete the operation. When the smoke became light and blue, the job was done. The air was completely blocked off to extinguish the fire and the heap left to cool. When it was torn apart, much of the wood was now porous black sticks, mainly of carbon, with all volatile components driven off by the heat. The charcoal was nearly pure carbon, so it would burn without smoke or yellow flame, and not contaminate anything with impurities. The use of charcoal as a fuel is discussed under Fuels. A typical hardwood charcoal was 20 lb to the bushel.
While the pile was burning, a watery liquid would trickle out of the bottom. This was pyroligneous acid, 80%-90% water, but including formic and acetic acids, methyl alcohol, formaldehyde, acetone. Resin in the wood would give rise to turpentine and tar. The gas, of which some 8000 cuft were produced per cord of wood, was 59% CO2, 33% CO, 3.5% CH4, 3% H2, and the remaining 1.5% heavier vapors. These were valuable by-products, but only feeble efforts were made to save and purify them. Now, charcoal is made in retorts and these products are recovered.
What we now call coal was used in local areas from ancient times, and was considered a kind of inferior fuel that gave odoriferous smoke. It was generally called stone coal. It was used in forges, for industrial purposes, in the 13th century in Liège, an early center of ironworking. Its widespread use began in the 17th century in England. Coal from the Tyne valley and the coast of Northumberland began to be exploited in bell pits, carried out by women with baskets, and was sent to London by sea. At this time, London had exhausted the forests in all directions, and charcoal was scarce. In fact, the use of charcoal for smelting had been outlawed, sending smelting to far-away districts, and even overseas to North America. This was called sea-coal, and there are references to it in Shakespeare.
The red-hot charcoal combines with the oxygen according to 2C + O2 → 2CO, producing the colorless, odorless gas carbon monoxide. The carbon monoxide burns above the coal with a blue, almost invisible flame, according to 2CO + O2 → 2CO2. The fire is completely safe as long as it gets enough air, and that any little CO that happens to escape is removed by ventilation. Today's tightly-closed houses make indoor charcoal fires unadvisable, however. The carbon monoxide is a very important feature in metallurgical fires. In the blacksmith's forge, it makes the atmosphere in the fire reducing, which means that iron heated in it will not oxidize or rust rapidly. In the iron furnace, it combines with the ore to give metallic iron. The iron ore is mixed with limestone, so the lime combines with the silica in the ore to make a slag that melts at low temperature. When all the iron ore has been reduced to metallic iron by the carbon monoxide percolating through it, the iron and slag are removed as a spongy mass, which is then hammered to consolidate it and to drive out the slag. When it cools, it is reheated in the forge fire. This was the way iron was made from prehistoric times until the 19th century, by direct reduction in a charcoal fire. In the United States, it survived until the mid-19th century, until driven out by the more efficient blast furnace and puddling furnace. It was still used later in some places, such as Sweden, to make very pure iron, since the charcoal introduced no deleterious impurities.
Carbon dissolves in liquid iron. If the forge fire were blown too assiduously, and the sponge melted, the iron would absorb carbon from the charcoal, its melting point would drop, and the whole thing would form a molten ball. When this ball solidified, it was hard and could not be forged. Therefore, it was useless and the day's work was lost. In the 18th century, Abraham Darby II (1711-63) of Colebrookdale found that this disaster could be turned to advantage in making castings of the liquid iron, as people had long done with bronze, so it was not wasted after all. When the hot iron and carbon solution solidified, some of the carbon formed the white, hard substance Fe3C, cementitite, and most of the rest made flakes of graphite in the iron. This cast iron, an alloy of iron and carbon, has remained of great importance. Abraham Darby III erected the first iron bridge at Ironbridge in 1779 from cast iron, and it still stands.
The first Abraham Darby (1677-1717) introduced coke into metallurgy, substituting an abundant resource, bituminous coal, for a scarce resource, charcoal, in 1709. Coke is to bituminous coal as charcoal is to wood, and is equally pure. This prevented contamination of the molten iron with sulphur and phosphorus, which had defeated earlier attempts to use coal for smelting. Darby's innovation, however, was not generally accepted for many years afterwards. Coke is discussed at more length below under Fuels.
Since ancient times, carbon had deliberately been put into iron by packing the iron in graphite, and placing it in a clay crucible carefully luted shut. When heated for an extended period at red heat, the iron absorbed a little of the carbon, a few tenths of a percent, but this was enough to change its properties drastically. When heated, this iron could be forged, like normal iron, but when heated and plunged into water, it became extremely hard. The new substance was steel (which we have seen gave its name to diamond). This is called cemented steel from the process of heating it with carbon. There were many different processes at different times and places, and the whole thing was largely a mystery surrounding trade secrets. It was, however, the key to effective weapons, as well as to ordinary tools like knives and scissors.
Fuels
The word "fuel" comes by a tortuous route from Latin focus, "hearth," via focalia, "things pertaining to a hearth." We burn the fuel to heat us and our food. Practically all of our fuels are carbon-based, for reasons that we will discuss in what follows. First, we have to look at how to express certain magnitudes associated with fuels.
A typical effect of burning a fuel is heating of water, and "heat" is expressed in terms of this. The calorie is the heat required to warm a gram of water by 1°C. The Btu (British thermal unit) is the heat required to warm a pound of water by 1°F. The relation between C and F degrees, and grams and pounds, make 1 Btu = 252 cal. Heat, we know, is actually energy, and measurement of the amount of energy required to heat water gives 1 cal = 4.186 J, and 1 Btu = 778.26 ft-lb. A watt is a joule per second, and a horsepower is 550 ft-lb per second, or 745.575 W. Heat cannot be transformed entirely into work under normal circumstances. The fraction that is converted to work in an engine is its thermal efficiency, η. The thermal efficiency of an automobile engine is around 25%, while an electric power station can have an efficiency of as much as 40%.
In engineering calculations, gases can be described closely enough by the ideal gas law, pV = nRT, where p is the pressure, V the volume, n the moles of gas in the volume V, T the absolute temperature, and R a universal constant whose value depends on the units chosen for the other quantities. Absolute temperature is expressed in kelvin, K, with K = C + 273.15, or in rankine, R, with R = F + 459.69. The values 273 and 460 are close enough for government work. Standard conditions are 0°C or 20°C and one atmosphere. If p is in pascal and V in cubic meters, then pV is in joule. R is then 8.314 J/gmol-K. A gmol is a mass in grams equal to the molecular weight. We need C = 12, O = 16, H=1, S = 32, N=14 and air=28.97. A gmol of any substance contains the same number of molecules, about 6.02 x 1023, Avogadro's number. An atmosphere is 1.01325 x 105 Pa, or 760 mmHg. If we use atmospheres to measure pressure, and liters (1000 cc) to measure volume, then R = 0.082 l-atm/gmol-K.
In the fps system, volume is measured in cubic feet (cuft), and pressure in pounds per square inch (psi), or atmospheres, where 1 atmosphere = 29.97 inHg. Standard conditions are often taken as 60°F and 30 inHg. The mol is now the pound mole, lbmol (which contains 453 times more molecules than the gmol). Convenient values for the gas constant are R = 0.7302 cuft-atm/lbmol-R or 10.73 cuft-psi/lbmol-R. A lbmol of gas occupies a volume V = RT/p = 0.7302 x 520 = 378.9 cuft. From this, we can find the mass per cuft of any gas under standard condtions, if we know the molecular weight. Since the molecular weight of methane, CH4 is 16, it weighs 0.04214 lb/cuft under standard conditions.
The specific gravity of a solid or liquid is the ratio of the density to the density of water, at specified temperatures. The specific gravity of a gas is often the ratio of the density to the density of air under the same conditions. For liquid fuels, the specific gravity is given in degrees API, where °API = 141.5/spgr - 131.5. 10°API corresponds to spgr = 1.0, and the higher the number of degrees, the smaller the specific gravity. 40°API corresponds to spgr = 0.825.
To begin our consideration of fuels, let's first look at hydrogen, which is not a commonly-used fuel but is often suggested as one. Hydrogen burns in air according to 2H2 + O2 → 2H2O, releasing 61,100 Btu/lb of heat if we condense the resulting water to a liquid, 51,623 Btu/lb if we do not. It takes 1050 Btu to vaporize a pound of water, and sometimes this energy is used, sometimes it is not. Every hydrogen-containing fuel has a high-heat value, where the water is condensed, and a low-heat value, where it is not. This is a nuisance in describing things, but it is easy to understand. A cubic foot of hydrogen under standard conditions contains 0.00264 lb, so burning it gets us about 161 Btu. A cubic foot of methane, the major constituent of natural gas, yields about 1000 Btu per cubic foot. Hydrogen power is not dense. It would be rather uneconomic to transport it by pipeline, or to store it in tanks.
Hydrogen forms an explosive mixture with air in the proportions by volume from 4% to 74.2%. The similar limits for gasoline vapor are 1.5% to 7.5%, for propane 2% to 10%, for methane 5% to 15%. This makes hydrogen an extreme explosion hazard. City gas used to contain hydrogen, and every winter city blocks would blow up here and there when the smallest leaks occurred. Hydrogen cannot be used as fuel for internal combustion engines, since it will not burn nicely--its antiknock properties are zero. It has been used as rocket fuel, however. It is expected to make a fuel cell fuel, but most small fuel cells burn things like alcohol, not hydrogen. I do not know if a reasonable hydrogen fuel cell has been devised. For all these reasons, hydrogen would not seem to have great promise as a general fuel.
There is another little drawback to hydrogen. There are no hydrogen mines. Most hydrogen on the planet is in the form of water--that is, it is already burned. To produce hydrogen, we have to put in more energy than we will receive by burning it. The economics of doing this with "free" solar energy are not good--the investment is large, and the hydrogen produced is small. In fact, the most economical ways to produce hydrogen at present use carbon fuels, by reforming hydrocarbons or by de-oxidizing water with hot carbon. Hydrogen at best is only an energy distribution system, not a prime energy source, and a rather inconvenient and dangerous system at that.
Solar energy is the source of most of the available energy on earth. It can be used directly to heat water, a use that is quite practical, if not responsive to variable demands. Sunlight can produce electricity with photovoltaic cells, which is also practical but expensive, and involves the inconveniences of storing electricity. Solar energy has been used for millennia, however, in the form of charcoal. Trees grow in the sun, then the wood is converted to charcoal, which is nearly pure carbon in a porous and easily used form. Charcoal is burned according to C + O2 → CO2, making available about 14,000 Btu/lb. Energy can be stored in the form of wood, and used when necessary, not at the constant rate of direct solar energy.
Human population has already far outstripped charcoal resources. Forest competes with arable land, and is disappearing steadily, in place of growing or remaining constant. As early as the 16th century, wood shortages were felt in the vicinity of cities (then much smaller than today's). The recourse was to stored forms of solar energy that were laid up over millions of years, but could be exploited in centuries. These finite reserves have supported the economy that has now grown to huge dimensions, and is using the reserves at a rapid rate. These fuels, which I shall call carbon fuels to spread the net wide, contain mainly carbon and hydrogen, with smaller amounts of oxygen, sulphur and nitrogen. The carbon and hydrogen burn as described above, with a small deduction for the cost of separating them. Sulphur contributes only about 4000 Btu/lb, from the reaction S + O2 → SO2. Fortunately, the amount of sulphur in carbon fuels is small, at most a few percent. Oxygen represents a part of the fuel that is already burnt, in effect, while nitrogen and what makes up the ash is inert.
The composition of a carbon fuel is given by a proximate analysis in terms of fixed carbon, volatiles, ash and moisture, or by an ultimate analysis of the dry fuel in terms of percentages of C, H, S, O, N and ash. The heating value of the fuel can be calculated in terms of the ultimate analysis. The Dulong equation is Btu/lb = 145.5C + 620(H - O/8) + 41S, while the Vondracek equation is Btu/lb = (160.5 - 0.112C')C + 486(H - O/10). The letters represent the percentages in the ultimate analysis, and C' is the percent carbon in a solid fuel. The heating value of a liquid petroleum fuel can be estimated by Btu/lb = 18,250 + 40 x °API.
Fuels are conveniently classified as solid, liquid and gaseous. Solid fuels are sold by weight, liquid and gaseous fuels by volume. We have already discussed charcoal. The other principal solid fuels are coal and coke. Coal is classified by rank. Anthracite contains more than 86% fixed carbon. It is hard, black and dustless. Bituminous either contains more than 69% fixed carbon, or has a fuel value of more than 11,000 Btu/lb. Sub-bituminous has a fuel value of 8300-13000 Btu/lb. Lignite, or brown coal, has a fuel value of less than 8300 Btu/lb. These ranks display less and less metamorphosis of the coal by heat and pressure. Sub-bituminous and lignite coals are subject to weathering, that is, disintegration on being exposed to the weather where they are subject to alternations of wet and dry. Bituminous and anthracite coals are not subject to weathering.
Many bituminous coals are called agglomerating, or coking coals because they swell upon heating, and then melt to produce a sintered cell structure, which forms a coherent coke. There is no necessary relationship between rank and agglomerating nature. Coals of lower rank, especially those that weather and contain pyrite, are subject to spontaneous combustion when stacked in a heap so that heat cannot escape. This is largely a function of the surface area presented for oxidation, so need not be feared for lump coal of high rank. Lump bituminous coal has an average density of about 50 lb/cuft, which includes the voids.
There are four main coal substances in bituminous coal. Vitrain is shiny and glassy, but very opaque. Durain is a dull, grey substance, hard and tough. Clairain is finely laminated vitrain and durain. The bright coal, vitrain and clairain, are separated by lenses of durain. These parts of the coal are clean and durable, but fusain is black and powdery, and responsible for all the dirt and dust. Banded bituminous coal, the common variety, consists of layers of vitrain and fusain. Splint coal is dull, consisting mainly of durain, and does not swell upon heating. The special coals cannel and boghead have unusually large amounts of volatile matter, but are noncoking. Cannel coal was made from pollen, boghead from seaweed. Boghead coal gives large amounts of tar and oil when distilled. Anthracite coal contains no fusain, so it is clean and burns with little smoke. However, anthracite is very difficult to light, while steam coal is much easier to get burning. The structure of coal is very complicated, and has not been well-deciphered. It is predominantly aromatic in structure, giving aromatic hydrocarbons upon distillation. The heating value of bituminous coal ranges from 10,500 to 14,400 Btu/lb.
Anthracite coal made an excellent stove fuel, better than charcoal, because it was denser and cleaner. It was the fuel of choice on the east coast of the U.S. until the coming of natural gas, and the exhaustion of the anthracite fields. It is difficult to light an anthracite fire unless you have experience with it. Kindling wood is required (liquid lighters will not suffice), and a significant amount of time. Once lighted, however, the fire would be steady and hot, with no smoke or fumes. Proper venting was required to avoid the carbon monoxide danger, as with charcoal. A good fire also requires equal-sized lumps. Mine-run coal was crushed (called breaking) and screened. Bone was material from the mine with 40% to 75% fixed carbon, slate with less than 40%. Both were eliminated from stove fuel, which was carefully graded in uniform sizes. Particles smaller than 1/32", called culm, were not sold and were used for industrial purposes, such as locomotive and boiler fuel. Culm was a necessary consequence of breaking, and a large amount resulted. Culm is still being economically recovered from waste heaps. Anthracite was discovered in America near Pottstown, Pennsylvania in 1790 by hunter Necho Allen, but it was quite a few years before its proper use was understood, around 1808. The use of anthracite in iron smelting was first tried in 1795, but this was not perfected until considerably later.
An excellent fuel for metallurgical purposes is coke, made by driving off the volatile matter from bituminous coal. Coke can be made like charcoal, in beehive coke ovens, so-named because of their shape. The idea is to burn the volatile matter in the coal to drive out the volatile matter. When the smoke at the chimney turned from thick and brown to thin and blue, the coke was made. The oven was cooled and the coke scraped out. The gases given off were so valuable (and so noxious) that by-product ovens were made like coal gas retorts (see below), and the volatiles condensed and used.
Coking coal swells when heated as the volatiles are driven off, creating a strong, cellular structure. Coke is a dull dark gray to a light silver gray, much unlike coal, and is a kind of artificial anthracite, almost pure carbon. To be useful in the blast furnace, it must be strong to keep the charge permeable to gases. In Britain, railway locomotives were required for some years after 1830 to burn coke, so they would not emit smoke. Coke was a good, but rather expensive, fuel. A typical foundry coke might be 90.5% fixed carbon, 8.64% ash, with less than 1% left of the volatiles. Coke has a heating value of about 13,000 Btu/lb, being nearly pure carbon. The purification resulting from coking prevents impurities from entering the iron, especially sulphur and phosphorus.
Liquid petroleum, or crude oil, can be used as a fuel as it comes from the ground. Its value and utility are greatly enhanced, however, by refining. This is, essentially, fractional distillation. The crude oil is heated gradually in a still. The most volatile components come off first, and are condensed. The temperature is raised, and the less volatile components are condensed. This continues as the temperature is raised, until the still contains only a thick black tar, the "bottoms." Each component can be characterized by the temperature range over which it is collected. Present-day refining is not limited by the initial contents of the crude oil, since heavy components can be cracked into lighter components, and lighter components reformed into heavier.
Petroleum naphtha, or gasoline (a Standard Oil trade name) came off from 38°C to 200°C. It is about 85% C, 15% H, and has a heating value of 18,900 Btu/lb. Its specific gravity is 55° to 70° API (0.7587-0.7022), so it weighs about 6 lb per gallon. This straight gasoline has an ignition temperature of 350°C, and requires about 14-15 lb of air per lb of fuel to burn completely. It is about 78 octane. There was originally little use for this fraction, and it was burned around the refinery or flared. The worst antiknock behavior is exhibited by straight-chain paraffinic hydrocarbons. The octane rating can be improved with branched chains, double bonds, or aromatic ingredients.
The next fraction, coming off between 160°C and 300°C, was called kerosene ("wax oil") by Standard Oil, and was the principal desired product, a fuel for lamps. To be safe in this use, the oil at normal temperatures should not have a high enough vapor pressure that a match would light the vapor before it reached the wick, possibly causing an explosion. A specification called the flash point was established to ensure safety. The flash point of a liquid fuel is the temperature below which the vapor will not catch fire under certain standard conditions. Kerosene must have a flash point above 73°F. In fact, it is usually from 110 to 130°F. Kerosene is a little heavier than gasoline, 40° to 48° API, about 6.7 lb per gallon. The heating value is the same as for gasoline, 18,900 Btu/lb. Kerosene is called "paraffin" in England, which was another trade name, this time of a firm that made it from shale oil in Scotland.
It is inevitable that some refiners would increase profits by putting as much naphtha in their kerosene as they could get away with. The casualties from exploding lamps caught national attention, and provoked legislation. Standard Oil bragged that their kerosene was kept to a Standard, and this was actually more or less true, helping to lead the company to success (along with some other less creditable reasons). Gasoline has a flash point of -47°F, far below normal room temperatures, so gasoline is always ready to explode, which is necessary for its use in engines. This is an excellent reason for not using gasoline in lamps that are not specifically made for it, or for using gasoline as a dry-cleaning fluid, or for other household uses that may suggest themselves.
The heavy oils coming off after kerosene could be used as fuels or as lubricants. They made good lubricants, and were more pleasant than the animal oils and greases previously used. Fuel oils are now denoted by number. No. 1 fuel oil is little different from kerosene, while No. 6 is heavy and viscous, requiring preheating so it will flow. The flash points run from 100°F to 150°F. The pour point is the temperature at which the oil will flow more or less readily. These are the distillate fuel oils, the best of the sort. residual fuel oils are what is left in the still when the other fractions have been boiled off. They are likely to be black with suspended carbon, and solid at room temperature. crude fuel oils are crude oils unsuited to refining. Fuel oil has the advantage of being much easier to store and handle than coal, and is a very desirable fuel.
Crude oil and liquid fuels are sold in the United States by the barrel (bbl) of 42 gallons, about 5.6 cuft or 158.57 litres. This is the old wine barrel, and just happened to be the size of barrel that was around when the first drilled wells in northwestern Pennsylvania began production in the 1860's.
A liquid carbon fuel that does not come from petroleum is ethanol, C2H5OH, which can be made organically by the fermentation of sugar produced by the hydrolysis of starch, processes catalyzed by enzymes. Ethanol provides 12,770 or 11,585 Btu/lb, about 2/3 that of petroleum. As a motor fuel, it has the advantage of excellent antiknock properties. Special engine designs are not required if it is used as a gasoline supplement. A useful blend is 65% gasoline, 20% ethanol, and 15% benzene. The benzene is to prevent separation if water gets into the mixture. This blend has a heating value of 17,224 Btu/lb, about 91% of that for straight gasoline. Benzene itself, which can be derived from coal, is a good motor fuel, providing 17,480 Btu/lb (low) with good antiknock properties. In German, Benzin is the word for motor fuel.
A liquid fuel that is used as a gas is LPG, liquefied petroleum gas, often propane, C3H8. Its boiling point is -43.8°F, so it liquefies easily under reasonable pressure, and there is no trouble with vaporization when it is used. Its specific gravity is 0.508, so it weighs 4.23 lb per gallon. The vapor is 1.522 times heavier than air, and 36.5 cuft are obtained for each gallon of liquid. The heat of vaporization is 183 Btu/lb, and the heating value is 21,690 Btu/lb, or 2514 Btu/cuft, twice that of natural gas. Burners must be adjusted to allow for this difference of heating value. Propane requires 15.6 lb of air per lb of fuel. The explosive limits are roughly 2% to 8%, making it safer than natural gas. Propane and butane can be used in internal combustion engines with complete success. LPG is a very convenient fuel, worth its premium price.
Gaseous fuels are easily made from coal, coke or oil. The iron blast furnace is a copious source of gas. It is blown to burn the carbon in its charge of coke, iron ore and limestone, producing CO that reduces the iron to metal. The gas that issues from the top of the stack has a lot of nitrogen and carbon dioxide, but usually about 30% CO that gives it a heating value of 90-110 Btu/cuft. This blast-furnace gas is used to heat the air for blowing the furnace, to heat reverberatory furnaces, and for other purposes around the steel works. A mixture of steam and air can be blown continuously through a bed of glowing coke to make producer gas. A typical producer gas might be 27% CO, 12% H2, 2.5% CO2 and 55% N2, with a heating value of 120-180 Btu/cuft. Producer gas is a clean fuel for heating pottery kilns, glass furnaces and similar facilities.
Blue gas or water gas is, however, gas that may be sold for use outside the factory. It is made by blowing air and steam alternately through hot coke. The "blow" with dry air brings up the temperature of the coke to white heat. Then the "run" with steam takes advantage of the reaction H2O + C → H2 + CO to make a gas that is typically 50% H2 and 40% CO, with 10% nitrogen and carbon dioxide, which are low because no air is used in the run phase. The heating value of this gas is 285-310 Btu/cuft. It burns with a blue flame, which gave it its name. An illuminating gas, one that burns with a luminous yellow flame, can be made by carburetting the water gas by adding the gases made by thermal cracking of a fuel oil. This was the gas normally supplied as city gas in later years. The high CO content made it possible to commit suicide by sticking one's head in the oven, but this no longer works, since natural gas is not poisonous. This is also the cheapest way to produce hydrogen gas for a hydrogen economy.
The original gas distributed for street lighting and home use from early in the 19th century was coal gas, made by distilling coal in horizontal cast-iron retorts. All of the volatile ingredients are driven off when the coal is heated to about 900°C, leaving a desirable gas coke in the retort. The condensable fraction of the gas driven off yields ammonia, benzene, coal tar, creosote and many other valuable carcinogenic substances. The gas made is about 49% H2, 42% CH4, 6% CO, and the usual 10% nitrogen and carbon dioxide. It provided 520-575 Btu/cuft. A very similar gas comes from by-product coke ovens, where conditions are optimized for making metallurgical coke instead of gas. The appropriate fraction of the liquid output was used as a lamp fuel under the name of coal oil, a name that was transferred to kerosene in later years.
Gas producers using oil could be made smaller and more convenient for portable use. They used oil and steam, and the water gas dual process, high temperatures, and the cracking of the oil to smaller fragments, to make excellent illuminating gas. One example was Pintsch gas, which had a heating value of 1300 Btu/cuft. Pintsch gas was widely used for lighting railway carriages.
Natural gas, which is largely methane, CH4, gives 1100-1150 Btu/cuft when pure. Other light hydrocarbons are often present, and enhance the heating value of the gas. It burns with a blue flame, so is not an illuminating gas. What light there is comes mainly from C2 molecules and the molecular fragments from the chain reaction. Most of the radiation is invisible. Methane is also called marsh gas, from its occurrence in marshes, were it burns as the Will-o'-th'-Wisp, and fire damp in coal mining, where it causes explosions. (Some say the Will o' th' Wisp is impure phosphine burning.) The discovery of the Welsbach mantle of cerium oxide around 1896 gave an excellent light, as good as that of an incandescent lamp, when heated with a nonluminous flame. Carburetion was removed from city gas when the mantle became common, and since then house gas has burned with a blue flame, like straight water gas. Natural gas, of course, does not need to be manufactured. It comes out of the ground when you punch a hole in the right place. Because natural gases from different sources have different amounts of carbon dioxide and so different heating values, gas is sold by heating value, not by volume. A therm is 100,000 Btu, or about the heating value of 100 cuft, called CCF in the trade. An MCF is 1000 cuft. The volumes are figured at standard conditions.
Another curious gas is acetylene, C2H2. It can be made as required by dropping H2O on CaC2, calcium carbide. Acetylene burns with a bright yellow luminous flame. Acetylene lamps using carbide were once common as bicycle lights and helmet lights for miners. They included a steel wheel and "flint" like a cigarette lighter, so all you needed was a can of carbide and a little water. They were so excellent that I am surprised they are no longer common. Carbide is formed by heating lime and coke in an electric arc furnace: CaO + 3C → CaC2 + CO. Acetylene is formed in the reaction CaC2 + 2H2O → C2H2 + Ca(OH)2. The slaked lime did make a bit of a mess if you weren't careful. The explosive limits of acetylene are 2.5% to 80%, so it is as bad as hydrogen in this respect. Acetylene burns with a hot blue flame when pre-mixed with oxygen. The oxyacetylene torch is used for welding and other purposes, and the acetylene is supplied in tanks. It is dissolved in acetone, since if you just tried to pressureize the tanks, the acetylene would blow up.
Buckyballs and Nanotubes
Buckminsterfullerene is the carbon molecule C60. It is a convex polyhedron with 12 equal pentagonal faces and 20 equal hexagonal faces, each pentagon surrounded by 5 hexagons. A model is shown at the right (see References). Its symmetry is icosahedral, point group Ih, with 12 five-fold axes, 20 3-fold axes, 15 2-fold axes and 15 planes of symmetry, of order 120. This group has irreducible representations of dimension 5, 4, 3 and 1, which will be the degeneracy of states of the corresponding symmetries. It has the same symmetry as the common football (soccer ball), and is nicknamed the "buckyball." The fascinating story of its discovery can be read in Baggott. A plane can be tessellated with regular hexagons, but a sphere requires the addition of pentagons, as was shown by Euler. He showed that F + V = E + 2, where F = number of faces, V = number of vertices and E = number of edges. For buckminsterfullerene, V = 60, F = 32 and E = 90.
Buckminsterfullerene is named from its resemblance to the "geodesic domes" designed by R. Buckminster Fuller (1895-1983), of which a famous example was the U.S. Pavilion at the Montreal World's Fair in 1967. He began to design domes in the 1920's, but not until after the 1947 patent did he concentrate on them. Over 2000 have been built. The domes are assembled from tetrahedrons and octahedrons of lightweight material, and generally have hexagonal faces, but pentagonal faces are there.
There are two classes of carbon-carbon bonds in C60, 30 connecting the pentagons to the hexagons, and 60 forming the pentagons. The first are 0.1386 nm long, the rest 0.1434 nm long. The average is 0.142 nm, exactly the bond length in graphite, where all the bonds are equivalent. Since each carbon forms three bonds, there is one spare electron per carbon to resonate around the rings, stabilizing them, as in graphite. The diameter of the molecule is 0.71 nm. If we assume the radius of a carbon is 0.07 nm, then 54% of the volume (about 0.19 nm3) of C60 is void. A saturated hydrocarbon, C60H60, a "fuzzy buckyball," can be formed by hydrogenation.
There are higher fullerenes, such as C70, C76, C78 and C84, composed of hexagons and pentagons with various symmetries. Fullerenes have been made inside fullerenes, so that they are concentric. Nested fullerenes can be made that resemble onions 50 nm in diameter, with 70 shells. In the work of synthesizing these fullerenes, needles were noticed that were found to be composed of nanotubes. These are helical assemblies of graphite hexagons. If the pitch is just right, the hexagons fit together smoothly (you might investigate this by rolling up paper with a hexagon net drawn on it). These were first seen in 1991, but similar structures had been noticed as early as 1960 by Roger Bacon at Union Carbide, also produced in an electric arc with carbon electrodes, when studying the much larger carbon whiskers that have found use in composite materials. Ijima, the 1991 discoverer, was the first to recognize that they were closed, smooth tubes, not rolled-up graphite.
The ends of the tubes are capped by spherical surfaces containing the necessary 12 pentagons, 6 at each end, while the tubes themselves are made only of hexagons. The tubes may be multiple, one inside the other. They are only a few nm in diameter (2-20 nm is usual), from which they acquired their name. They are, as may be expected, extremely strong--stronger than carbon fibres or any other known material, for their size. Their growth apparently starts from a pentagon in one cap as one of the helical (chiral) fullerene structures, and then finds it easier to continue as tube of hexagons when the equator is reached. Buckyballs, buckytubes and buckyonions show that graphite would rather not be flat when it is in small amounts. All of the structures avoid the energetically unfavorable dangling bonds at the edges of a sheet, because they have no edges.
Buckyballs and nanotubes are much more interesting than they are useful, but ingenious uses may yet be found. Buckyballs can clathrate (jail) things, and nanotubes can conduct electricity and contain metal fillings. These properties may lead to applications. They were detected in the interstellar medium, produced by red giant carbon stars, which send soot into space. In fact, the study of interstellar absorption led to the discovery of buckyballs. These interstellar buckyballs seem to clathrate lanthanum.
Radiolarians are microscopic animals of the plankton that live in silica geodesic domes composed of hexagons and pentagons, and the occasional square, which greatly resemble fullerenes. Reticulum plasmatique and Aulonia hexagona are illustrated in Baggott. The pseudopoda extend from the openings to feed. A hexagonal net can tessellate a bathroom floor, but if it is to form a closed surface, pentagons must be introduced, as Euler proved.
References
E. H. Kraus and C. B. Slawson, Gems and Gem Materials, 5th ed. (New York: McGraw-Hill, 1947). Everything about diamonds.
L. Pauling, General Chemistry (New York: Dover, 1970). Stereograms of diamond and graphite structures, p. 169.
J. K. Salisbury, Kent's Mechanical Engineers' Handbook, 12th ed., Power Volume (New York: John Wiley & Sons, 1950). Section 2, Fuels.
J. Baggott, Perfect Symmetry (Oxford: Oxford University Press, 1994). The story of buckminsterfullerene. The radiolarians are on p.82, a buckyball is on p.71, and the Montreal dome is on p.58.
A buckyball model kit can be obtained from ScienceStuff. The model is 37 cm in diameter, and quite impressive. Assembling it, which takes about two hours, teaches the structure very well.
L. Pauling and R. Hayward, The Architecture of Molecules (San Francisco: W. H. Freeman, 1964). Excellent illustrations of many structures, including graphite and diamond.
Pictures of diamond and graphite kindly furnished by Amethyst Galleries, Inc.. These images are copyrighted, but can be used noncommercially. This is an excellent website and specimens can be purchased online. This is probably the best mineral website, and the company should be supported for making it available.
A. Holmes, Physical Geology 1st ed.(New York: Ronald, 1945). Chapter XVI.
A. Holmes, Physical Geology 2nd ed.(New York: Ronald, 1965). pp. 273-276.
Diamonds is an excellent Australian website on diamonds.
E. L. Quinn and C. L. Jones, Carbon Dioxide (New York: Reinhold, 1936).
Composed by J. B. Calvert
Created 9 December 2002
Last revised 11 September 2006


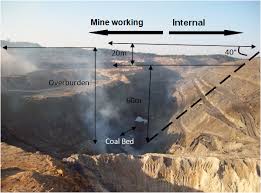
Comments
Post a Comment